mRNA Folding Mechanisms and Approaches at the Secondary Structure Level
A review of RNA folding tools and an intuitive way to explain Minimum-Free Energy Concepts

RNA Energy Dot Plot Intro and Replicate for Folding Software
RNA sequences fold into secondary and tertiary conformations. These folds are driven by nucleotide base pair affinity - G like C, A likes U. You may know of them from DNA as Watson Krick Base Pairs.
Each nucleotide binds with one partner at a time, where long stretches where they have binding partner are more favourable than shorter, closer binding partners in terms of the sequence. Often, you may see that coding sequences on an mRNA molecule only fold at their tails while the open frame has a stem structure.
However, some folds still do occur. In fact, when base-pair complementarity is not present, we get these loops and hairpins across the elongated structure. This, in culmination, produces these secondary structures which play key roles in RNA molecule function [analogous to the features of a protein].
More commonly, secondary structures are highly conserved throughout function or rapidly changing under given circumstances which influence the hydrogen bonds between the base pairs.
For example, riboswitches have regulatory open elements which, when in the presence of other organic molecules, breaks bonds and thus forces the RNA molecule to change structure [the goal of this writing is to give you a better intuitive understanding for this process].

These riboswitches are at the end of a traditional mRNA molecule, and play a significant role in regulating protein expression at the ribosomal subunits. For mRNA molecules, this folding can be extrapolated with a bit of understanding on biology and access to compute power.
This is part 1 of a 2-part series which explains the rationale behind my current mRNA folding optimizer project, and its potential to expand our immunorepertoire.
Brief Introduction to the Kinematics of Folding
We can think of selective RNA folding as an environment where evolution favours the dominant traits. In a not so similar but very subtle way, our body has synthesized base pairs such that it generates competition across base pairs to fold based on a variety of factors.
Primarily, we’ve thought of available energy and how we can create the most efficient molecules, but these need to be balanced with functionality [i.e. translation rate of information exchange], the pH, temperature, and its ability to interact with the rest of the body [needs to degrade via enzymatic reactions from Ribonuclease]. In some cases, the structure can be optimized to fold such as certain codons align with other codons and typically form complementary strains that are just long, double-stranded RNA folds [duplex RNA].
Available chemical energy and potential is the primary factor that influences the conformation of mRNA molecules. We call the lowest-energy conformation the Minimum-Free-Energy Conformation.
We can go beyond this and describe RNA molecule arrangement as probabilistic to the variety of factors and how this probabilistic model helps explain the ability for a single molecule to transition between different optimal structures.
By now, you like me, would have realized the significance of this property for the RNA World hypothesis and even the role it plays as a diverse, robust biological machine.
Before we jump into how these probabilistic models are designed, it’s better to familiarize yourself with a simplified notation of RNA secondary structure. In bioinformatics, most conformations are recorded in Energy Dot-Plot Format.

A dot-plot is really just a way for us to figure out which base pairs with which. The diagonal acts as our axis/current base pair. We read the dot plot by traversing the diagonal and looking at whether at its x coordinate, there is a base pair formation.
These are indicated by the coloured energy states, and different coloured energy states indicate different folding conformations. If an energy state is at the intersection of the horizontal position of a base pair, the y-coordinate of the energy state aligns to another nucleotide at the diagonal. Therefore, the relative positions of each energy state correspond to the base pairing of that structure.
As a result, you can see some patterns. For one, stem structures have a straight diagonals and all gaps represent some form of loop. As we go through this section, you’ll notice that interior loops, for example, will only have 1 missing energy state between a stem structure.

Loops ultimately mean that base-pairs at the x-coordinate have no binding partners so they branch away and become independent [often yields loop]. If you notice, spaced out stems are considered as branches.
Types of Secondary Structures and Embeddings
Before we jump into the secondary sub-structures commonly found in RNA molecules, it is important to recognize the notation that we can use to better fit these structures into a give context.
A-A-A-A-A-C-C-C-C-U-U-U-U-U
0-1-2-3-4-5-6-7-8-9-10-11-12-13
i < k > jWe can provide indexes for each base in a sequence and denote the 5’ as i with 3’ as j. Most algorithms, which we will be going through soon, typically find substructures by starting in the middle with 2 indices and pushing each index outwards [start from centre of diagonal on dot plot].

Notice that j and i start at different values, and we start at (4,6) rather than (0,9). k represents whether a pairing occurs in a given range after each iteration, but know that this notation will be commonly used throughout.
1. Stem
More than one base pair appears in the form of a group of contiguous base pairs. This resulting structure motif is described as a stem.
A good example of the structure format is provided in the figure below.

At the secondary level, stem motif appears as flat but can always twist and turn at the tertiary level.
##A stem structure is determined by the base pair between i, j and those below/above it.
GGCAUGCU
i j
...
A-U -> a stem is determined by whether 2 consecutive base
C-G pairs are formed
G-C i+1 = G, j-1=C
G-U i = G, j = U
...
In a similar fashion, the energy required for a conserved stem motif is calculated by the current and previous base pairs to 1) validate that it really is a stem structure & 2) include the biological significance of stems being more conserved assuming that nearby structures are less random/more stable
eS(i,j) = turner_energy((bp[i], bp[j]),(bp[i-1], bp[j-1]))2. Hairpin
Two complementary sequences [stems] are joined by non-pairing bases which are in the form of a loop [no bonds] .
Each hairpin ultimately act as a little loop at the end of a given stem and closes it off, so the 5’ → 3’ direction is still conserved by the stem position.
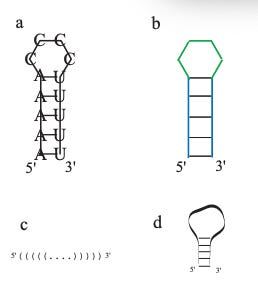
At i, j [A-U] right before the hairpin, the loop starts to form from base pairs who cannot bond together. A neater visualization is also provided in the form of Dot-Bracket Notation.
Here, you can connect this back to the previous sort of i→k←j diagram and the energy dot plot where each time an energy state is selected, it creates this sort of triangular bridge that shows the pairing of 2 bases. In a similar way, dot-bracket notation simplifies this idea where base pairs whose position [i+n] coincide with a given “(“ pairs with base at position [j-n] that coincides with “)”, and “.” represents base pair k that remains unpaired.
This is why loops are formed in the first place since every base pairs with another base at the opposite/reflective position in the sequence. The idea of symmetry may help with intuition here.
Going back to our initial example of "AAAAACCCCUUUUU", the dot-bracket notation of this sequence indicates that CCCC will not bond due to lack of complementarity.
##Because of lack of base pairing between C-C, no bonds are formed but loop structure is still conserved.
GGCAUGCU
i j
...
C≠C -> Since they can't pair, they align side by side and
C≠C form loop as 5'...C-C-C-C...3'
...3. Internal
An internal loop is a product of unpaired bases on 2 antiparallel strands in the stem structure. Between these two stems with unpaired bases in between them, they form bulges on both sides.

We refer to an internal loop for symmetrical break loops between the stem where the number of bases on each side/side of the stem are equal. A bulge is then really just an unpaired
n1, n2 represent both sides of the loop at 5' and 3'. The idea of a bulge and internal creates 3 secondary substructure possibilities:
Possibility 1:
G-C
n_1a - - n2_a ]- 2 unpaired bases on both sides of the stem
n_1b - - n2_b ∴ n1 == n2
U-A
Possibility 2:
G-C
n_1 - - ]- extra base pair on n1 side creates right-bulge
G-C ∴ n1>0,n=0 only unpaired on 5' strand [i=i'+1]
G-U
U-A
Possibility 3:
G-C
- - n2 ] - extra base pair on n2 side creates left-bulge
G-C ∴ n1=0,n2>0 only unpaired on 3' strand [j=j'+1]
G-U
U-A4. Multi-branch loops
This secondary structure is really considered to be a generalization of sorts for more complex loops in an mRNA molecule. Typically, these look like junctions whose branches have the prior substructures.

Each multi-branched loop consist of several stem-loop type structures, and so while most other loops are easy to detect just by traversing a RNA sequence from both ends and then inward, multi-loops become a bit more complex.
range of base-pairs in a multi-branched loop consist of pairs
(i1,j1), (i2,j2), (ik, jk) with k base-pairs.
If 3 or more stems branch off of a substructure, we can consider the unpaired region to be a multi-branched loop where pairs (ia,ja), (ib,jb), (ic,jc)We’ll go back into why this is, but know that we really only have 2 de-facto RNA folding algorithms: Nussinov’s and Zuker’s algorithms. Both of these have subtle differences which will help Zuker’s account for more realistic folds in comparison to Nussinov’s.
Conventions in Stability based on Minimum Free Energy
To find the free energy of a secondary substructure, we can decompose each individual loop to evaluate stability in terms of energy. We do this at each base pair in substructure that closes a loop and then use a different set of rules to evaluate the stability of the loop.
Tools like ViennaRNA can easily determine the energy states by summing all total energy states for all kinds of loops and structures [we’ll be jumping into this soon].
Going back to our original model, stability of an mRNA transcript is determined by the presence of sequences within an mRNA known as cis-elements. However, this just means that the molecule is harder to denature and thus can survive and function in highly volatile conditions.
We can use certain parameters that allow you to sum up the relative stability of each nearest-neighbour pair in a given duplex.

These are all thermodynamic parameters where you can look at all possible 2-nucleotide pairs and observe their relative stability. For stems, their energy is dictated by the stacking of neighbouring base-pairs.

Most computational algorithms used for Minimum Free Energy calculate the value (q) in terms of the Boltzmann weight of the free energy.

such that Free Energy (E) is in terms of β

The gas constant R and temperature T are measures that are used with the Boltzmann weight (β) to relate the average relative kinetic energy of particles in a gas with thermodynamic temperature of the gas-evaluating translation factors for mRNA.
If we can minimize this function for the free energy of a given sequence s for all P folding conformations, we can find the optimal secondary structure to maximize stability.
The stability can also be used for insights on RNA-protein interactions with respect to how the ribosome is reading the mRNA molecule, whether the free-energy conformation is effective for max translation, and other interaction factors with enzymes and such.
Free energy’s biological advantage is in the ability for the mRNA molecule to be detected. This is the distinction between a cognate (shared structure/affinity) vs. non-cognate mRNA molecule. If the mRNA molecule is non-cognate, the low affinity between the ribosome and molecule introduces more errors in recognition and thus the accuracy and effectiveness of the translation step.
Let 'c' be cognate frequency, 'nc' be non cognate frequency
∆∆𝐺𝑏𝑖𝑛𝑑 = ∆𝐺𝑏𝑖𝑛𝑑𝑐-∆𝐺𝑏𝑖𝑛𝑑𝑛𝑐 which is related to the populations or dissociation constants:
∆∆𝐺𝑏𝑖𝑛𝑑 = −𝑅𝑇 ln(𝑃𝑐/𝑃𝑛𝑐) = −𝑅𝑇𝑙𝑛(𝐾𝑑𝑛𝑐/𝐾𝑑𝑐)
We define excess free energy difference between two states as ∆𝐺 = −𝛽^−1𝑙𝑛 (𝑍𝐵/𝑍𝐴) = −𝛽^−1𝑙𝑛〈exp(−𝛽∆𝑈)〉A
To simply these definitions, we can look into laws of thermodynamics as a TLDR.
Ultimately, the question we’re trying to answer is why does energy flow in certain directions in ways? Well, we describe this in the form of Gibbs Free Energy, a definition for a system under constant pressure.
The formula for Gibbs Free Energy is G = H − TS where H is the measure of enthalpy [potential to do work], T is the absolute temperature and S is the entropy.
Change in enthalpy is also defined as change in heat. If a reaction gets an increase of kinetic energy in reaction A + B = C, the change in enthalpy for C needs to have more energy.
Gibbs Free Energy = Change in Enthalpy of the Reaction - (Temperature * Change in Entropy)
Change in enthalpy = heat of reaction
When your reactants have more energy than products, enthalpy is negative to indicate spontaneous reaction, and vice versa [decrease in enthalpy is an exothermic reaction]
Change in entropy = heat-transfer to melt ice * temperature
increase in entropy of surroundings is an exothermic reaction
Refer to index for calculations of observed free energy with respect to individual types of secondary structures
ViennaRNA, a very popular RNA folding software, uses Gibbs Free Energy in relation with the Nearest Neighbour Method. Here, we consider Gibbs Free Energy (G) as all available energy. The goal is to find mRNA structure whose hydrogen bonds’ energy requirements are below or equal to the Minimum Gibbs Free Energy.
Therefore, G is function of total energy of a system [H] and entropy [S] and temperature of system [T].
Enthalpy - what happens when system is in movement? A lot of the potential energy is turned into kinetic energy. Therefore, Total energy [H] or enthalpy has decreased since there is less potential energy after each second in the reaction. In the case of biology, potential energy is the hydrogen bonds or base-pairing bonds between the different nucleotide base pairs. Entropy - when we increase disorder, typically referring to spontaneous reactions -> the random movement of all particles with respect to particlesTemperature - if you have increased temperature, you're ultimately increasing entropy due to movement of moleculesTherefore, a greater temperature/entropy would reduce Gibbs Free Energy, and a greater enthalpy increases Gibbs Free Energy.
If there is less Gibbs Energy, it indicates that we have more entropy and less enthalpy, indicating that a spontaneous reaction has just occurred as there is more kinetic and less potential energy [Exergonic]
If there is more Gibbs Free Energy, temperature/entropy has decreased to reduce chaos and thus not start a spontaneous reactions [Endergonic]
If Gibbs Free Energy = 0, then there is equilibrium between entropy and temperature along with enthalpy [Equilibrium]
For example, in cellular respiration, we break bonds in glucose to release energy, this increases our kinetic energy and thus reduces Gibbs Free Energy, therefore exogenous reaction or releasing heat/energy. In photosynthesis, you are assembling energy through CO2 and water in the form of glucose or starch, therefore it is endogenous where Gibbs Free Energy is higher.
Our molecule’s Gibbs Free Energy *measured in kcal/mol) is based on bringing the total energy as low as possible.
If we were to plot this on a graph of free energy vs. reaction progress, we want the bottom-most corner to have the lowest free energy during the reaction progressing when it comes for the molecule to fold.
To be continued in Pt. 2 → We’ll be going over how Zuker and Nussinov’s Algorithms work + diving into some neat implementations. Until then, why don’t you check out some other stuff?